Coding Genotype Probabilities
The field names for marker information have to be constructed very carefully, because the core code uses these names to understand what data is being presented to it. Each field name has a number of sections, separated by underscores.
Mappings for Genetic Marker Probabilities
Section titled “Mappings for Genetic Marker Probabilities”Use these mappings to construct the marker header name for genotype probabilities:
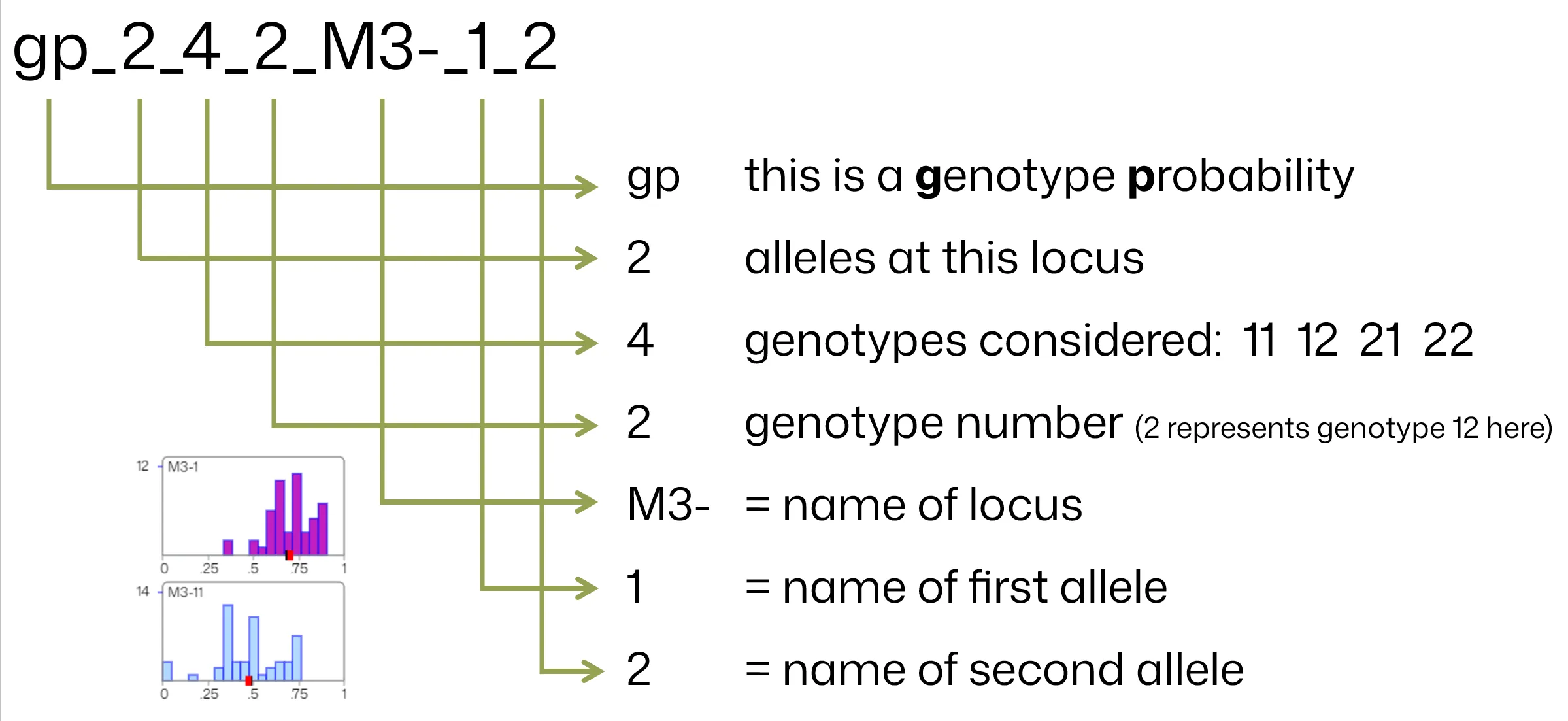
Component Details
Section titled “Component Details”The table below describes further each component:
| Component | Description |
|---|---|
| Type | ”gp” or “GP” for a genotype probability. Genotypes Probabilities have multiple fields/columns per marker as described below. |
| Number of alleles (nA) | Usually 2, but any number > 2 is accepted. |
| Number of genotypes | This is the number of genotypes that could be present, including lethals, even if not all genotypes are in your dataset. So if you have no 22 genotypes in your dataset, and you are grouping 12 and 21 together as one genotype, then these are the genotypes in this list: [11, (12 or 21), 22], and the number of genotypes is 3, not 4. |
| Genotype Number | This is only included if type is GP. This is the number of the genotype to which the probability belongs, as in the examples above for 2 and 3 alleles and the two possible numbers of genotypes for each of these. So for 2 alleles and 3 genotypes (2_3) the genotype numbers are 1 for p(11), 2 for p(heterozygote) and 3 for (22), and the data columns contain probabilities ranging 0 to 1. Multiple fields for a given marker should be grouped together in the data file, and presented sequentially according to genotype number. Genotype number 1 can be omitted, as noted below. |
| Marker/Locus Name | It is critical that the multiple genotype probability fields for a single marker locus use exactly the same marker name. It is recommended to append a dash to the marker name as in the “M1-” example above because marker name is usually followed directly by the name of one of its alleles in graphical output. Do not use a space in a name. Marker names should not be longer than 12 characters. |
| Allele 1 Name | Alphanumeric text. Do not use a space in a name. No longer than 4 characters. |
| Allele 2 Name | As above |
| Allele n name | As above, name all nA alleles |
For genotype probabilities, where the number of genotypes is n, there can be n fields for that marker, with values summing to 1 for each individual candidate (a checksum is carried out, with some allowance for rounding error), but it is also possible to omit the field for the first genotype, giving n-1 fields for the marker. In the latter case, the code detects the omission, and the probability for the first marker genotype is generated in the code. For example, two biallelic markers (M3 and M4) are presented below as genotype probabilities:
gp_2_4_1_M3-_1_2
gp_2_4_2_M3-_1_2
gp_2_4_3_M3-_1_2
gp_2_4_4_M3-_1_2
And
gp_2_4_2_M4-_1_2
gp_2_4_3_M4-_1_2
gp_2_4_4_M4-_1_2
For M3-, all four genotype probabilities are included, but for M4- the first probability is not included, such that it will be deduced by MateSel.
The core MateSel code converts all genotype and genotype probability information to handle genotype probabilities for all nA² genotypes at each marker locus. This is appropriate, as even where candidate genotypes are specified, the results for prospective progeny have to be given as probabilities (eg. a mating 12 x 12 gives progeny probabilities of ¼ for each genotype: 11, 12, 21, 22). Moreover, even if supplied candidate genotypes or genotype probabilities do not show direction of inheritance for heterozygotes (eg. 12 versus 21), such information can still be generated for prospective progeny (eg. progeny from a 11 x 22 mating are all 12, and none are 21).
Candidate Values
Section titled “Candidate Values”The content above just tells us how to construct the header name for genotype probabilities, but what values do we enter for each candidate? Each value will be a continuous number ranging from 0 to 1 representing the probability of the candidate having the genotype in question.
Missing Genotypes
Section titled “Missing Genotypes”Genotypes denoted as 0 or a period . are treated as missing. Where a full set of genotype probabilities is provided and they do not sum to 1 (strictly abs(sum-1.)>0.000001 ) then that locus is treated as missing for that individual. Where an incomplete set of genotype probabilities is provided (all but the first) and the first probability is deduced to be negative (strictly < -0.00001) then an error message will appear on the interface or console. An individual with missing genotypes or genotype probabilities is allocated the raw mean of genotype probabilities for current candidates (i.e. excluding those who are only listed as candidates in CommittedMatings.txt) that have legal genotypes or genotype probabilities.