Genomic Relationship Matrix (GRM)
A Genomic Relationship Matrix (GRM) among your candidates can be used to help manage coancestry and progeny inbreeding. As you supply the GRM, as described below, you can introduce any type of relationship matrix to MateSel by this method, whether or not it involves genomic information. See Meuwissen et al 2020 “Management of Genetic Diversity in the Era of Genomics” for a discussion on different types of GRM.
In addition, your GRM might include blends between pedigree and genomic relationship matrices, genomic relationship matrices pertaining to specific genomic region(s) (see “Target specific genomic region(s) for management of diversity and inbreeding” section below), and even matrices that include dummy individuals that represent groups of real individuals, and their predicted average relationships with all other candidates (see Handling large groups of individuals as single “dummy individuals”).
GRM File Format
Section titled “GRM File Format”Make a file called CandGRM.txt and fill it with GRM elements in one of three ways, as in the three following examples:
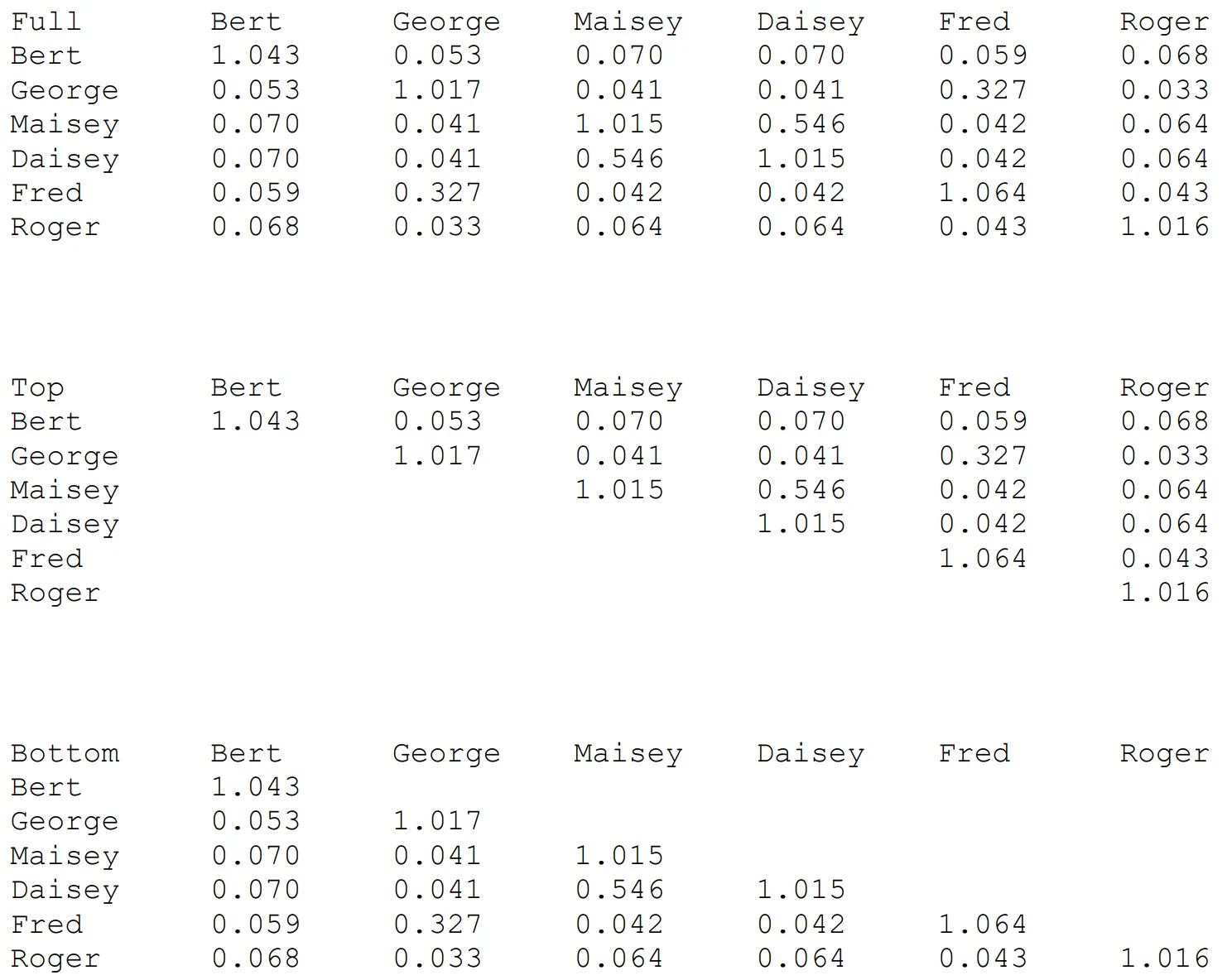
The Top-Left string in this file must be either “Full”, “Top” or “Bottom” (case sensitive), for a full-stored or half-stored (top or bottom) GRM. These files are read free-format, and do not have to be spaced as above, which is only done to help with illustration. GRM elements can have any number of decimal places shown. For your CandGRM.txt file:
- The individual IDs used (Bert, George, etc., in the examples above) should be exactly the same as for the corresponding individuals in the main datafile used for the run.
- The overall order of individuals is not important (George could come before Bert), but order within the file should be consistent – for example in the Full file, an individual should have the same row number and column number, giving a symmetric GRM.
- You can include individuals in the GRM that are not candidates and even not in the main data file.
- You can have individuals that are candidates missing from the GRM file (but see How Matesel handles candidates missing in the GRM supplied.
Using the GRM File
Section titled “Using the GRM File”Ensure your CandGRM.txt file is in the same directory as your main data file. When you open the data file in MateSel, you will be given options on how to use the GRM:
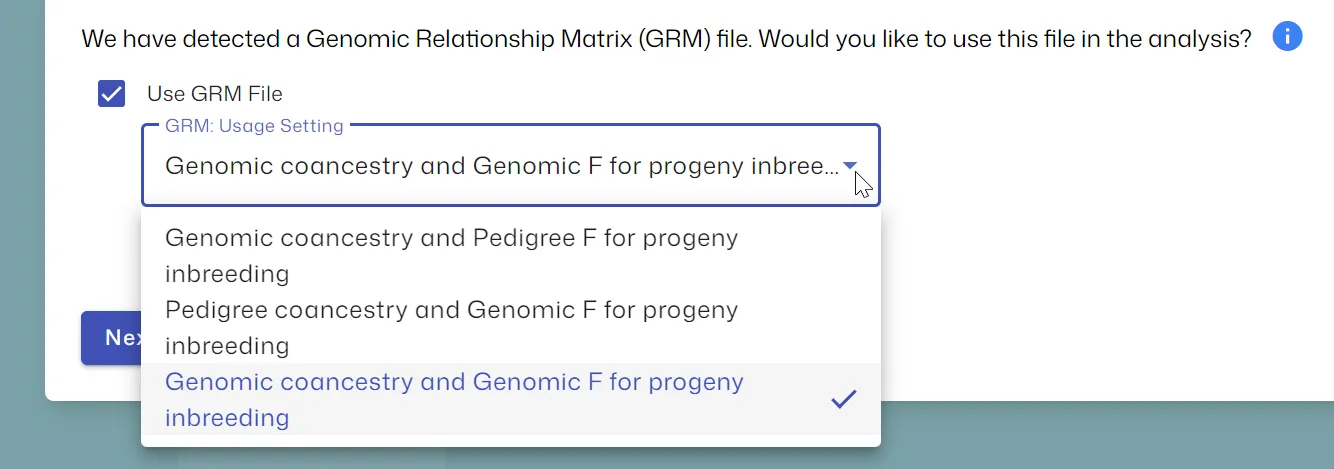
- Genomic coancestry and Pedigree F for progeny inbreeding (Internal Code Number: 1)
- Pedigree coancestry and Genomic F for progeny inbreeding (Internal Code Number: 2)
- Genomic coancestry and Genomic F for progeny inbreeding (Internal Code Number: 3)
Genomic coancestries and F values are calculated from your GRM elements, just as if they were NRM elements (ie. no scaling in MateSel).
With this method, it is possible for the one GRM file to remain unchanged over some time, across several MateSel runs. Each run cherry-picks what it needs from the file, and whatever elements are missing are imputed, if possible, within MateSel. Of course, the GRM file should be updated whenever new DNA info is available, or a new blending is made. But from a practical point of view, the mating process and the genomic information can be somewhat asynchronous. Of course this can all work with the updating of GRM or equivalent for the genetic evaluation – but it can be managed separately if required for some reason.
Note: Use of the GRM for genomic EBVs can take place prior to MateSel runs, by supplying gEBVs in the “Index” field.
How MateSel imputes missing relationships
Section titled “How MateSel imputes missing relationships”MateSel in fact works with missing GRM elements, whether they are missing in the file CandGRM.txt, or they are present in the file but set to a missing value of -99999.
The supplied GRM is first augmented by appending rows and columns for the missing candidates, and these rows and columns are populated with the missing value code of -99999.
The approach is to seek the four parents of the two individuals that each missing element relates to. For individuals 1 and 2 the parents are S1, D1, S2 and D2. The missing element is taken as the mean of the following four elements S1S2, S1D2, D1S2 and D1D2, if all four of these elements are present and not missing (Note that they can become non-missing following successful imputation).
The method progresses from ancestors to descendants, “painting” the full ordered-pedigree matrix from the top-left corner towards the bottom right corner, to impute parents before progeny. After a full pass seeking all 4 parental elements for each missing element, a full pass is made where at least 3 parental elements can be found, and the mean of these 3 taken to impute the missing element, then passes at 2 and 1 parental element.
Higher stringency was sought in a test dataset by resetting the minimum requirement to 4 parental elements after each new imputation of an element, and also by iterating this whole process. However, neither of these strategies increased the number and quality of imputations made.
Missing elements that cannot be imputed by this method are allocated a value equal to the mean element plus one unit of standard deviation among elements.
Here is the sort of output you will see on the console. This is from a test dataset with relatively sparse pedigree information available – another reason to use a GRM for coancestry and inbreeding management:

Target specific genomic region(s) for management of diversity and inbreeding
Section titled “Target specific genomic region(s) for management of diversity and inbreeding”Some concern has been voiced that selection involving major QTL will result in too much homozygosity or lack of diversity in the genomic regions surrounding them, and that steps should be taken to conserve that diversity. But there are potential benefits from doing the opposite, at least in the short term, by reducing diversity management in QTL regions to further promote their selection. (see: this article)
Controlled across specific genomic regions can be set up outside MateSel by preparing GRMs that are calculated with attention paid to specific region(s). It is likely that you will have a consensus GRM that has placed varying levels of emphasis on different regions across the genome. You can test the impact of using such a GRM as follows:
- Make a run using the consensus GRM and parameters/settings appropriate to your desires. Before stopping the run, give it a name e.g. “ConsensusGRM”.
- Now you can make different runs using GRMs that target specific regions, and then compare the saved consensus results into these runs to observe how the latter performs at each region of interest (see Compare Runs).
- You can also do this the other way around – observe how policies that target a single region (or a given set of regions) perform under the consensus GRM.